Extrapolated Species Richness in a Species Pool
specpool.RdThe functions estimate the extrapolated species richness in a species
pool, or the number of unobserved species. Function specpool
is based on incidences in sample sites, and gives a single estimate
for a collection of sample sites (matrix). Function estimateR
is based on abundances (counts) on single sample site.
Usage
specpool(x, pool, smallsample = TRUE)
estimateR(x, ...)
specpool2vect(X, index = c("jack1","jack2", "chao", "boot","Species"))
poolaccum(x, permutations = 100, minsize = 3)
estaccumR(x, permutations = 100, parallel = getOption("mc.cores"))
# S3 method for class 'poolaccum'
summary(object, display, alpha = 0.05, ...)
# S3 method for class 'poolaccum'
plot(x, ...)Arguments
- x
Data frame or matrix with species data or the analysis result for the
plotfunction.- pool
A vector giving a classification for pooling the sites in the species data. If missing, all sites are pooled together.
- smallsample
Use small sample correction \((N-1)/N\), where \(N\) is the number of sites within the
pool.- X, object
A
specpoolresult object.- index
The selected index of extrapolated richness.
- permutations
Usually an integer giving the number permutations, but can also be a list of control values for the permutations as returned by the function
how, or a permutation matrix where each row gives the permuted indices.- minsize
Smallest number of sampling units reported.
- parallel
Number of parallel processes or a predefined socket cluster. With
parallel = 1uses ordinary, non-parallel processing. The parallel processing is done with parallel package.- display
Indices to be displayed.
- alpha
Level of quantiles shown. This proportion will be left outside symmetric limits.
- ...
Other parameters (not used).
Details
Many species will always remain unseen or undetected in a collection of sample plots. The function uses some popular ways of estimating the number of these unseen species and adding them to the observed species richness (Palmer 1990, Colwell & Coddington 1994).
The incidence-based estimates in specpool use the frequencies
of species in a collection of sites.
In the following, \(S_P\) is the extrapolated richness in a pool,
\(S_0\) is the observed number of species in the
collection, \(a_1\) and \(a_2\) are the number of species
occurring only in one or only in two sites in the collection, \(p_i\)
is the frequency of species \(i\), and \(N\) is the number of
sites in the collection. The variants of extrapolated richness in
specpool are:
| Chao | \(S_P = S_0 + \frac{a_1^2}{2 a_2}\frac{N-1}{N}\) |
| Chao bias-corrected | \(S_P = S_0 + \frac{a_1(a_1-1)}{2(a_2+1)} \frac{N-1}{N}\) |
| First order jackknife | \(S_P = S_0 + a_1 \frac{N-1}{N}\) |
| Second order jackknife | \(S_P = S_0 + a_1 \frac{2N - 3}{N} - a_2 \frac{(N-2)^2}{N (N-1)}\) |
| Bootstrap | \(S_P = S_0 + \sum_{i=1}^{S_0} (1 - p_i)^N\) |
specpool normally uses basic Chao equation, but when there
are no doubletons (\(a2=0\)) it switches to bias-corrected
version. In that case the Chao equation simplifies to
\(S_0 + \frac{1}{2} a_1 (a_1-1) \frac{N-1}{N}\).
The abundance-based estimates in estimateR use counts
(numbers of individuals) of species in a single site. If called for
a matrix or data frame, the function will give separate estimates
for each site. The two variants of extrapolated richness in
estimateR are bias-corrected Chao and ACE (O'Hara 2005, Chiu
et al. 2014). The Chao estimate is similar as the bias corrected
one above, but \(a_i\) refers to the number of species with
abundance \(i\) instead of number of sites, and the small-sample
correction is not used. The ACE estimate is defined as:
| ACE | \(S_P = S_{abund} + \frac{S_{rare}}{C_{ace}}+ \frac{a_1}{C_{ace}} \gamma^2_{ace}\) |
| where | \(C_{ace} = 1 - \frac{a_1}{N_{rare}}\) |
| \(\gamma^2_{ace} = \max \left[ \frac{S_{rare} \sum_{i=1}^{10} i(i-1)a_i}{C_{ace} N_{rare} (N_{rare} - 1)}-1, 0 \right]\) |
Here \(a_i\) refers to number of species with abundance \(i\) and \(S_{rare}\) is the number of rare species, \(S_{abund}\) is the number of abundant species, with an arbitrary threshold of abundance 10 for rare species, and \(N_{rare}\) is the number of individuals in rare species.
Functions estimate the standard errors of the estimates. These only
concern the number of added species, and assume that there is no
variance in the observed richness. The equations of standard errors
are too complicated to be reproduced in this help page, but they can
be studied in the R source code of the function and are discussed
in the vignette that can be read with the
browseVignettes("vegan"). The standard error are based on the
following sources: Chiu et al. (2014) for the Chao estimates and
Smith and van Belle (1984) for the first-order Jackknife and the
bootstrap (second-order jackknife is still missing). For the
variance estimator of \(S_{ace}\) see O'Hara (2005).
Functions poolaccum and estaccumR are similar to
specaccum, but estimate extrapolated richness indices
of specpool or estimateR in addition to number of
species for random ordering of sampling units. Function
specpool uses presence data and estaccumR count
data. The functions share summary and plot
methods. The summary returns quantile envelopes of
permutations corresponding the given level of alpha and
standard deviation of permutations for each sample size. NB., these
are not based on standard deviations estimated within specpool
or estimateR, but they are based on permutations.
The plot function for accumulation models works both with
poolaccum and estaccumR. It draws the mean of
accumulations against the number of sites. ggvegan provides
function autoplot that can also show the empirical confidence
intervals of accumulations.
Value
Function specpool returns a data frame with entries for
observed richness and each of the indices for each class in
pool vector. The utility function specpool2vect maps
the pooled values into a vector giving the value of selected
index for each original site. Function estimateR
returns the estimates and their standard errors for each
site. Functions poolaccum and estimateR return
matrices of permutation results for each richness estimator, the
vector of sample sizes and a table of means of permutations
for each estimator.
References
Chao, A. (1987). Estimating the population size for capture-recapture data with unequal catchability. Biometrics 43, 783–791.
Chiu, C.H., Wang, Y.T., Walther, B.A. & Chao, A. (2014). Improved nonparametric lower bound of species richness via a modified Good-Turing frequency formula. Biometrics 70, 671–682.
Colwell, R.K. & Coddington, J.A. (1994). Estimating terrestrial biodiversity through extrapolation. Phil. Trans. Roy. Soc. London B 345, 101–118.
O'Hara, R.B. (2005). Species richness estimators: how many species can dance on the head of a pin? J. Anim. Ecol. 74, 375–386.
Palmer, M.W. (1990). The estimation of species richness by extrapolation. Ecology 71, 1195–1198.
Smith, E.P & van Belle, G. (1984). Nonparametric estimation of species richness. Biometrics 40, 119–129.
Note
The functions are based on assumption that there is a species pool: The community is closed so that there is a fixed pool size \(S_P\). In general, the functions give only the lower limit of species richness: the real richness is \(S >= S_P\), and there is a consistent bias in the estimates. Even the bias-correction in Chao only reduces the bias, but does not remove it completely (Chiu et al. 2014).
Optional small sample correction was added to specpool in
vegan 2.2-0. It was not used in the older literature (Chao
1987), but it is recommended recently (Chiu et al. 2014).
Examples
data(dune)
data(dune.env)
pool <- with(dune.env, specpool(dune, Management))
pool
#> Species chao chao.se jack1 jack1.se jack2 boot boot.se n
#> BF 16 17.19048 1.5895675 19.33333 2.211083 19.83333 17.74074 1.646379 3
#> HF 21 21.51429 0.9511693 23.40000 1.876166 22.05000 22.56864 1.821518 5
#> NM 21 22.87500 2.1582871 26.00000 3.291403 25.73333 23.77696 2.300982 6
#> SF 21 29.88889 8.6447967 27.66667 3.496029 31.40000 23.99496 1.850288 6
op <- par(mfrow=c(1,2))
boxplot(specnumber(dune) ~ Management, data = dune.env,
col = "hotpink", border = "cyan3")
boxplot(specnumber(dune)/specpool2vect(pool) ~ Management,
data = dune.env, col = "hotpink", border = "cyan3")
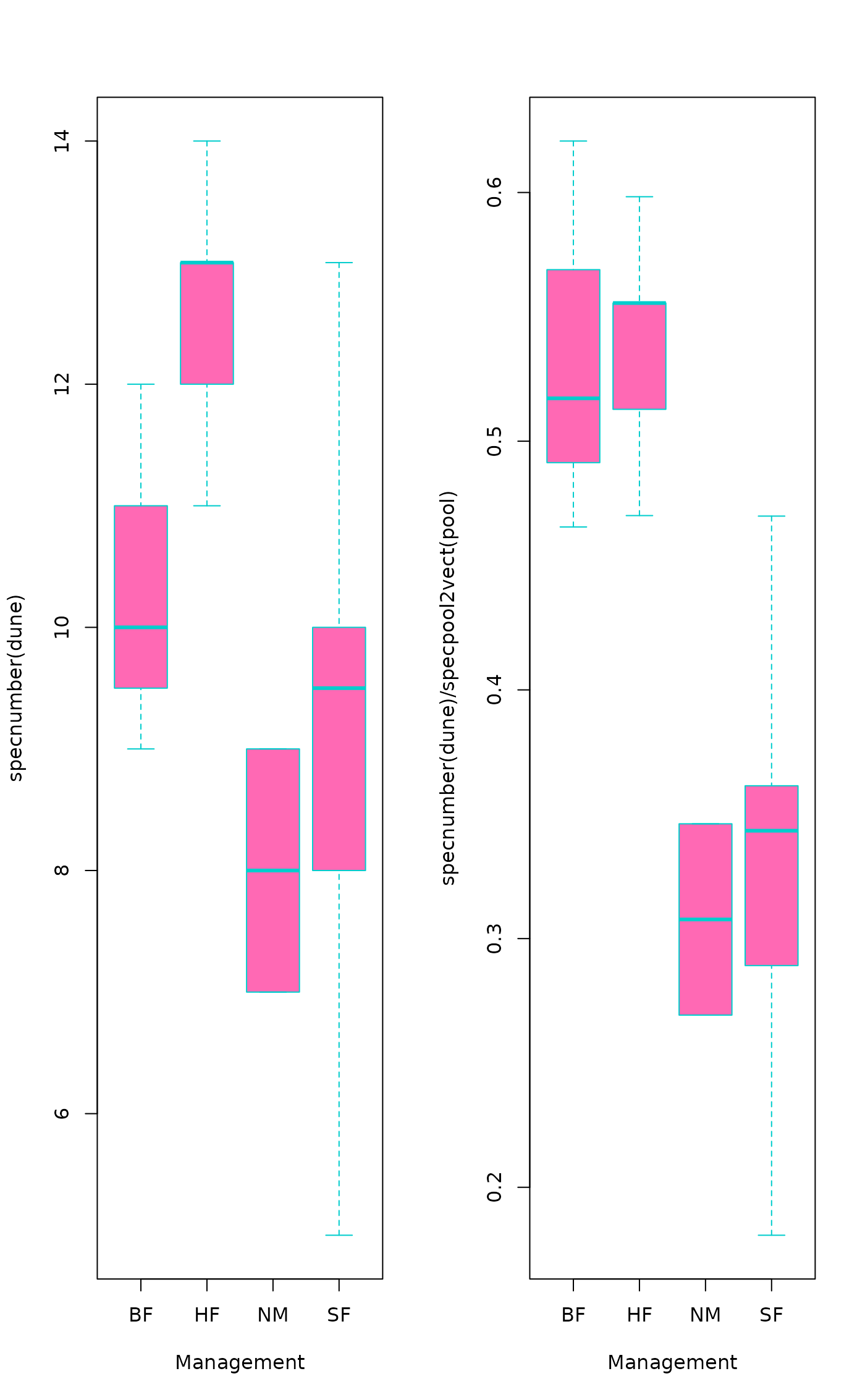 par(op)
data(BCI)
## Accumulation model
pool <- poolaccum(BCI)
summary(pool, display = "chao")
#> $chao
#> N Chao 2.5% 97.5% Std.Dev
#> [1,] 3 163.4563 136.5829 182.7655 12.004706
#> [2,] 4 176.5412 159.4328 195.6195 10.661510
#> [3,] 5 184.7591 165.7690 209.2479 12.627215
#> [4,] 6 190.2313 170.7455 216.4161 11.162749
#> [5,] 7 194.4566 175.7297 214.7811 10.517144
#> [6,] 8 199.2450 183.4371 224.8769 11.145997
#> [7,] 9 202.7078 187.0660 226.5574 12.142578
#> [8,] 10 205.7778 189.6709 229.1099 11.515657
#> [9,] 11 209.5071 191.9658 232.9952 12.109400
#> [10,] 12 211.9727 193.8087 237.8248 12.306252
#> [11,] 13 214.0954 194.9569 245.6762 12.220963
#> [12,] 14 216.2447 201.2563 246.5172 12.510035
#> [13,] 15 218.5823 202.5286 245.9757 13.227008
#> [14,] 16 219.1243 201.8979 243.1138 10.717775
#> [15,] 17 222.1347 203.7530 247.1392 11.678482
#> [16,] 18 223.1669 205.5961 250.0492 11.646743
#> [17,] 19 225.3469 204.9797 248.1653 11.825882
#> [18,] 20 226.6293 209.6916 253.6622 11.272303
#> [19,] 21 227.4581 211.3089 247.0192 10.265336
#> [20,] 22 227.9943 213.4813 249.1369 10.058572
#> [21,] 23 230.0601 211.7946 255.8265 12.296751
#> [22,] 24 231.5155 212.6402 254.0672 13.729715
#> [23,] 25 231.9116 213.3656 261.2438 12.560858
#> [24,] 26 233.0002 216.2592 261.8214 12.578114
#> [25,] 27 233.9318 215.6036 265.8193 12.089534
#> [26,] 28 233.9705 217.3018 265.1881 11.326573
#> [27,] 29 234.4725 215.9046 260.8103 10.671249
#> [28,] 30 235.2889 214.8192 262.3533 12.256566
#> [29,] 31 236.2261 215.3472 266.6174 12.410319
#> [30,] 32 237.0387 216.5141 261.1317 10.954032
#> [31,] 33 236.4132 216.7257 253.9088 10.154365
#> [32,] 34 237.0079 218.2307 257.6891 9.948949
#> [33,] 35 237.6703 219.5076 262.1426 10.520467
#> [34,] 36 237.7306 219.3470 258.2005 9.641592
#> [35,] 37 238.1567 220.5272 256.2735 8.935588
#> [36,] 38 239.0198 222.4706 260.1543 9.149970
#> [37,] 39 239.1022 223.2163 257.6613 8.116614
#> [38,] 40 238.8933 223.6656 255.6869 7.913056
#> [39,] 41 238.7612 224.1268 252.5317 7.482334
#> [40,] 42 239.1360 225.4690 255.3129 7.897372
#> [41,] 43 239.0753 226.4815 257.2072 7.975106
#> [42,] 44 238.9184 226.9847 257.8264 7.713831
#> [43,] 45 238.3132 227.0721 256.3079 6.883987
#> [44,] 46 238.2723 229.2662 253.6119 6.035264
#> [45,] 47 237.4575 229.2711 251.0192 5.188615
#> [46,] 48 236.9787 228.9313 247.8913 4.152311
#> [47,] 49 236.5269 233.7865 242.9539 2.327362
#> [48,] 50 236.3732 236.3732 236.3732 0.000000
#>
#> attr(,"class")
#> [1] "summary.poolaccum"
plot(pool) ## use ggvegan::autoplot to show CI envelope
par(op)
data(BCI)
## Accumulation model
pool <- poolaccum(BCI)
summary(pool, display = "chao")
#> $chao
#> N Chao 2.5% 97.5% Std.Dev
#> [1,] 3 163.4563 136.5829 182.7655 12.004706
#> [2,] 4 176.5412 159.4328 195.6195 10.661510
#> [3,] 5 184.7591 165.7690 209.2479 12.627215
#> [4,] 6 190.2313 170.7455 216.4161 11.162749
#> [5,] 7 194.4566 175.7297 214.7811 10.517144
#> [6,] 8 199.2450 183.4371 224.8769 11.145997
#> [7,] 9 202.7078 187.0660 226.5574 12.142578
#> [8,] 10 205.7778 189.6709 229.1099 11.515657
#> [9,] 11 209.5071 191.9658 232.9952 12.109400
#> [10,] 12 211.9727 193.8087 237.8248 12.306252
#> [11,] 13 214.0954 194.9569 245.6762 12.220963
#> [12,] 14 216.2447 201.2563 246.5172 12.510035
#> [13,] 15 218.5823 202.5286 245.9757 13.227008
#> [14,] 16 219.1243 201.8979 243.1138 10.717775
#> [15,] 17 222.1347 203.7530 247.1392 11.678482
#> [16,] 18 223.1669 205.5961 250.0492 11.646743
#> [17,] 19 225.3469 204.9797 248.1653 11.825882
#> [18,] 20 226.6293 209.6916 253.6622 11.272303
#> [19,] 21 227.4581 211.3089 247.0192 10.265336
#> [20,] 22 227.9943 213.4813 249.1369 10.058572
#> [21,] 23 230.0601 211.7946 255.8265 12.296751
#> [22,] 24 231.5155 212.6402 254.0672 13.729715
#> [23,] 25 231.9116 213.3656 261.2438 12.560858
#> [24,] 26 233.0002 216.2592 261.8214 12.578114
#> [25,] 27 233.9318 215.6036 265.8193 12.089534
#> [26,] 28 233.9705 217.3018 265.1881 11.326573
#> [27,] 29 234.4725 215.9046 260.8103 10.671249
#> [28,] 30 235.2889 214.8192 262.3533 12.256566
#> [29,] 31 236.2261 215.3472 266.6174 12.410319
#> [30,] 32 237.0387 216.5141 261.1317 10.954032
#> [31,] 33 236.4132 216.7257 253.9088 10.154365
#> [32,] 34 237.0079 218.2307 257.6891 9.948949
#> [33,] 35 237.6703 219.5076 262.1426 10.520467
#> [34,] 36 237.7306 219.3470 258.2005 9.641592
#> [35,] 37 238.1567 220.5272 256.2735 8.935588
#> [36,] 38 239.0198 222.4706 260.1543 9.149970
#> [37,] 39 239.1022 223.2163 257.6613 8.116614
#> [38,] 40 238.8933 223.6656 255.6869 7.913056
#> [39,] 41 238.7612 224.1268 252.5317 7.482334
#> [40,] 42 239.1360 225.4690 255.3129 7.897372
#> [41,] 43 239.0753 226.4815 257.2072 7.975106
#> [42,] 44 238.9184 226.9847 257.8264 7.713831
#> [43,] 45 238.3132 227.0721 256.3079 6.883987
#> [44,] 46 238.2723 229.2662 253.6119 6.035264
#> [45,] 47 237.4575 229.2711 251.0192 5.188615
#> [46,] 48 236.9787 228.9313 247.8913 4.152311
#> [47,] 49 236.5269 233.7865 242.9539 2.327362
#> [48,] 50 236.3732 236.3732 236.3732 0.000000
#>
#> attr(,"class")
#> [1] "summary.poolaccum"
plot(pool) ## use ggvegan::autoplot to show CI envelope
 ## Quantitative model
estimateR(BCI[1:5,])
#> 1 2 3 4 5
#> S.obs 93.000000 84.000000 90.000000 94.000000 101.000000
#> S.chao1 117.473684 117.214286 141.230769 111.550000 136.000000
#> se.chao1 11.583785 15.918953 23.001405 8.919663 15.467344
#> S.ACE 122.848959 117.317307 134.669844 118.729941 137.114088
#> se.ACE 5.736054 5.571998 6.191618 5.367571 5.848474
## Quantitative model
estimateR(BCI[1:5,])
#> 1 2 3 4 5
#> S.obs 93.000000 84.000000 90.000000 94.000000 101.000000
#> S.chao1 117.473684 117.214286 141.230769 111.550000 136.000000
#> se.chao1 11.583785 15.918953 23.001405 8.919663 15.467344
#> S.ACE 122.848959 117.317307 134.669844 118.729941 137.114088
#> se.ACE 5.736054 5.571998 6.191618 5.367571 5.848474